Research Overview
My work centers around the physiology, genomics, and evolution of rhizosphere bacteria.
Evolution of pathogenic/beneficial lifestyles and rhizosphere fitness
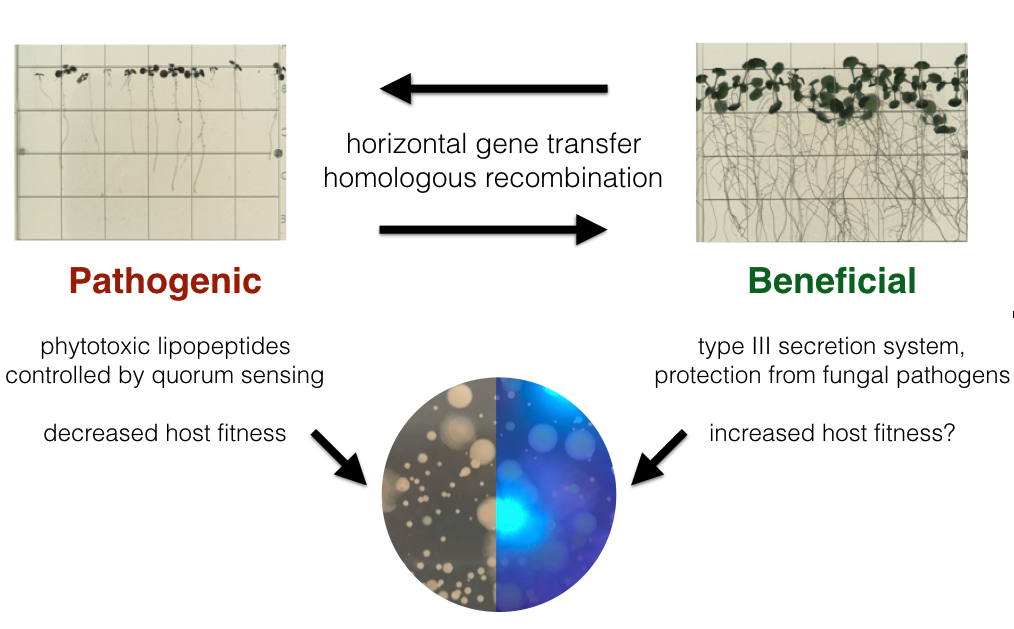
A major research interest of mine is how elements of the core and flexible genome interact to shape bacterial fitness in the rhizosphere, especially in the context of host health. Within the Pseudomonas brassicacearum clade there are strains that are both pathogenic and beneficial in the rhizosphere. In previous work, I have found that convergent evolution of the pangenome through gain and loss of genomic islands allows closely related P. brassicacearum strains to transition from commensal to pathogenic lifestyles in the rhizosphere. Pathogenicity is largely determined by the presence a pathogenicity island encoding a quorum-controlled phytotoxic lipopeptide biosynthesis cluster which I characterized using genetic and genomic methods.
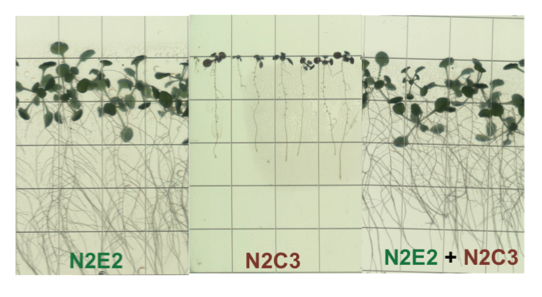
Moving forward, I am interested in uncovering the genetic basis of commensal behavior in this clade. Commensal strains have a conserved set of features including a type III secretion system, an antifungal biosynthetic cluster, as well as pathways for modulating the phytohormones auxin and ethylene. I am using genetics and in planta experiments to understand the effects these pathways have on various plant hosts. Interestingly, beneficial strains are capable of protecting Arabidopsis seedlings from the effects of pathogenic strains. To understand the basis of this protection, I am using LacZ-based fitness assays, transposon screens, and genetics. More broadly, using simplified synthetic communities from this clade might lead to better understanding of fitness during rhizosphere colonization.
I have assembled a collection of 33 strains with sequenced genomes from the P. brass clade isolated from a variety of plant-associated environments, including 26 isolated by Dr. Kelly Craven from switchgrass at the Noble Foundation. P. brass strains are generalists, capable of colonizing a variety of plant-associated environments. However, together they share a pangenome of over 9,000 genes, indicating a great deal of genotypic plasticity. To understand how the pangenome might shape fitness in varying plant environments, I plan to grow the entire population of 33 strains on a panel of diverse plants and assess strain fitness using a technique called FREQ-seq. By analyzing the strains enriched under a single condition, I will identify shared genes which could illuminate the basis of host adaptation within a clade of rhizosphere bacteria assumed to be generalists.
Nutrient niches in the rhizosphere
Sequencing of the conserved 16S ribosomal rRNA gene has revealed that the plant rhizosphere contains thousands of distinct bacterial species. The principle of competitive exclusion implies that there are subtle differences in the niches each of these strains inhabit, otherwise coexistence would be unstable. Microbiome studies have revealed that niche differentiation might occur spatially (e.g. rhizosphere vs. endophytic compartment), or temporally during plant host development. One understudied parameter that likely underlies niche differentiation is the use of plant-derived nutrients.
Different bacteria in the rhizosphere likely utilize different plant-derived nutrients. To identify these preferences, I am using RB-TnSeq libraries I constructed in several well-studied plant-associated bacteria, including Pseudomonas spp., Herbaspirillum seropedicae SmR1, Paraburkholderia phytofirmans PsJN, and Sinorhizobium meliloti 1021. By combining saturating transposon mutagenesis with next-gen sequencing, it is possible to ascertain the fitness of individual mutants under various conditions in a genome-wide manner. Using this technique on bacteria grown alone or in simple communities in the presence of plant root exudate will reveal how taxonomically divergent bacteria partition plant-derived nutrients to structure plant-associated communities.
Nitrogen cycling in agricultural soils
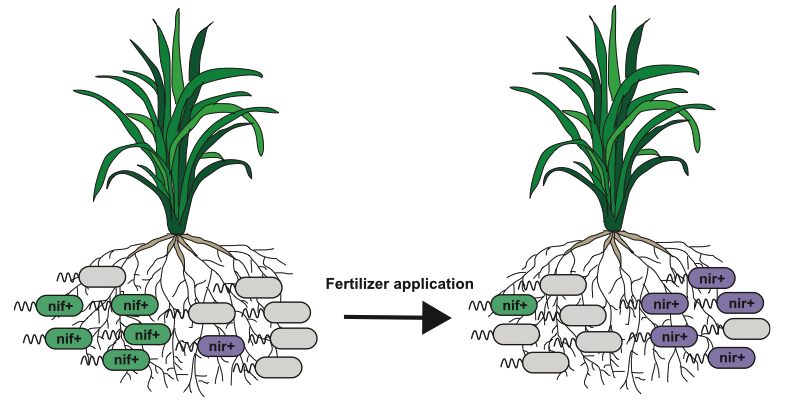
The widespread adoption of synthetic nitrogen fertilizer (SNF) for agriculture during the 20th century is a central pillar of the green revolution that has dramatically increased crop yields. However, current SNF utilization is ecologically unsustainable due to substantial greenhouse gas emissions and severe eutrophification of rivers, lakes, and oceans. In the absence of SNF, a major input source of nitrogen in agricultural systems comes from a bacterial process known as biological nitrogen fixation (BNF) . While BNF has long been studied in bacteria that form root nodules in certain plants (mostly legumes), graminaceous plants can also form associations with BNF-capable bacteria that provide fixed nitrogen to the plant (i.e “associative” BNF). These associative BNF bacteria are increasingly touted as a possible sustainable alternative to SNF for providing fixed nitrogen to cereal crops. However, because BNF requires a substantial amount of energy, it is unclear how an associative BNF strain would benefit from fixing nitrogen in excess of its own cellular demands. Furthermore, because small amounts of ammonia or nitrate stifle BNF in model nitrogen-fixing bacteria, it seems unlikely that plant-associated bacteria would fix nitrogen in all but the most nitrogen-limited settings. These evolutionary concerns are obstacles to using associative BNF strains as inoculants for enhancing crop nutrition, since selection and competition would seem to favor loss or inactivation of BNF.
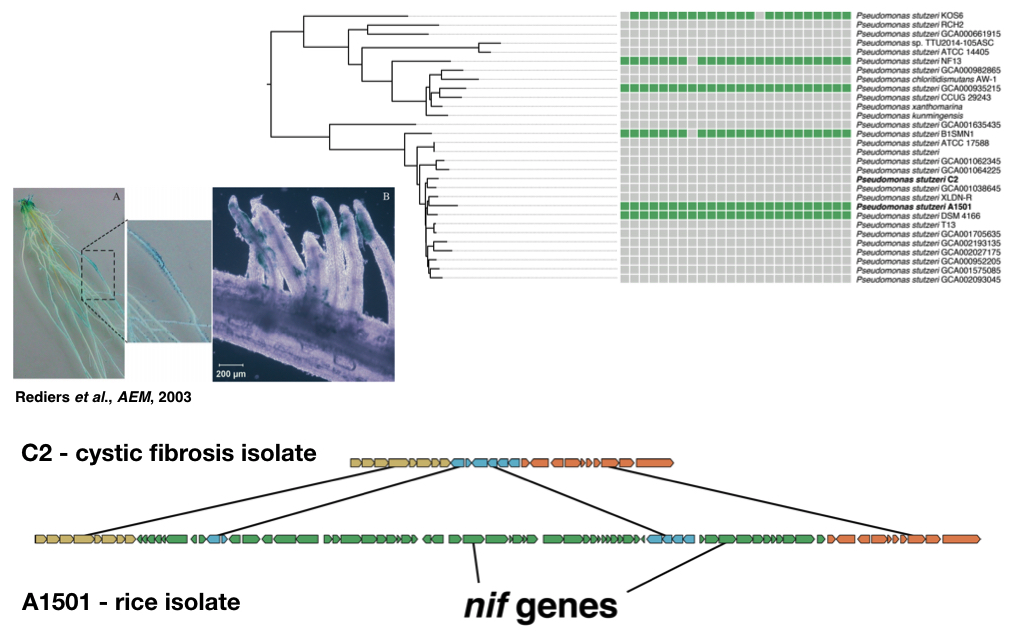
In a survey of published genome sequences from several associative BNF groups (Herbaspirillum, Burkholderia, and Pseudomonas stutzeri), I have found that the core nitrogen fixation (nif) genes are frequently gained and lost over short evolutionary distances, suggesting that associative BNF may only be adaptive under certain conditions. Moreover, it means that inferring the presence of nif genes from 16S data alone is impossible, even for well-studied model BNF taxa. To understand how associative BNF impacts fitness within bacterial populations, I am opting for a “top-down” approach using amplicon sequencing of rice-associated communities grown in the greenhouse on collected soils treated with different fertilization regimens. By varying the form and amount of exogenous nitrogen, I will ascertain the effect that this important environmental parameter has on the structure of the rice microbiome and the relative abundance of known BNF bacteria. However, since closely related strains can vary in their nif gene content, I have also developed a direct isolation methodology for isolating nitrogen-fixing bacteria. Ultimately, I will identify populations of closely related bacteria that contain both nif+ and nif- individuals. By studying these bacteria using comparative genomics, genetics, and gnotobiotic plant assays, I will understand how BNF impacts bacterial fitness in plant-associated niches and what genomic features and pathways are associated with the nif genes. In the future, I hope to extend this experimental framework to other agricultural model systems, to understand more broadly how associative BNF functions in different plant host contexts.